FDA査察の種類、観察事項分類(VAI,OAI,NAI)について
FDA(アメリカ食品医薬品局)は、医薬品等がFDAの規制に適合しているかどうかを確認するために製造業者に対してcGMP査察を行います。FDAの査察は主に以下のとおり分類できます。
- For-Cause Inspection(原因査察):
- 特定の問題やクレーム、事故が発生した場合、FDAはその原因を特定するために製造業者を査察することがあります。
- Pre-Approval Inspection(事前承認査察):
- 新しい医薬品や医療機器が市場に導入される前に、その製造施設が製品を製造するための適切な条件を備えているかどうかを確認する査察です。これは新薬の承認前に行われます。
- Routine Surveillance Inspection(通常の監視査察):
- 定期的に、製造業者や輸入業者の施設を査察し、製品の品質や規制の遵守状況を確認する査察です。
- Compliance Follow-up Inspection(遵守フォローアップ査察):
- 過去に規制違反があった場合、その状況が改善されているかどうかを確認するための査察です。
- Surveillance Inspection after Recall(リコール後の監視査察):
- 製品のリコールが行われた場合、その後の製造プロセスや品質管理の改善状況を確認する査察です。
また、FDA査察結果は観察事項の内容に基づき、以下のとおりVAI、OAI、NAIに分類されます。
OAI、またはVAIとなった場合は、引き続きFDAと連携しながら改善に向けた対応を行う必要があります。
なお、FDAはGMP適合性調査証明書等のCertificationは発行しておりません。
- VAI (Voluntary Action Indicated) - 自主的対応推奨: →Form483発行
- VAI (Voluntary Action Indicated) とは、FDAが査察結果に基づいて何らかの是正措置を講じることを勧告する場合に使われる分類です。企業はFDAの指摘事項に対して自主的に対処し、改善措置を実施することが期待されます。
- 通常Form483発行が発行され、期限までに回答します。
- OAI (Official Action Indicated) - 公式措置必要: →Form483発行+Warning Letter発行
- OAI (Official Action Indicated)とは、FDA査察官で重大な規制違反や安全性の懸念を発見し、何らかの是正措置を講じることを勧告するのみならず、警告書を発行することによりFDAが公式な措置を講じる可能性がある場合に使用される分類です。これには、製品の差し止め、製造許可の取り消し、法的な手続きの開始などが含まれます。
- 通常、Form483が発行された上で、Warning Letterが発行されます。ただし、Form483の回答内容によってはWarning Letterが回避される場合もあるようです。
- NAI (No Action Indicated) - 措置不要:
- これは、FDAが査察で特に問題が見つからなかった場合に使われる分類です。製造業者が規制遵守に合格し、適切な品質管理プロセスを実施していると判断された場合に適用されます。
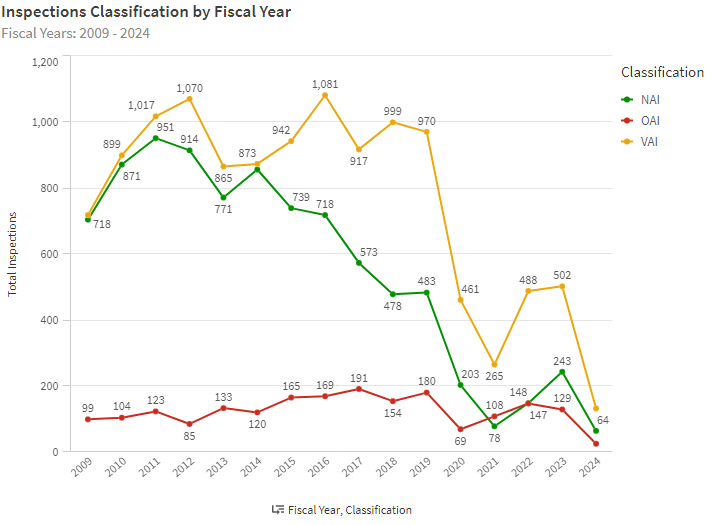
参考までに2009年から2024年までに実施したFDA査察(医薬品のみ)のNAI、OAI、VAIの割合は以下のとおりです。